H and H2#
import profess
import ase_tools
import matplotlib.pyplot as plt
import numpy as np
$H$ atom#
The hydrogen atom potential is $v(\mathbf{r})=-\tfrac{1}{r}$ in atomic units. Solving the associated Schrödinger equation yields the ground state energy $E_0=-\tfrac{1}{2}$.
For a single, isolated electron, we may infer its wave function from the electron density, $\psi(\mathbf{r}) = \sqrt{n(\mathbf{r})}$. Furthermore, its kinetic energy is given exactly by the Weizsäcker functional,
$$ T_W[n] = \int \mathrm{d}\mathbf{r} , \sqrt{n(\mathbf{r})} \left(-\frac{1}{2}\nabla^2\right) \sqrt{n(\mathbf{r})}. $$
For this reason, we can obtain $E_0$ as a simple example of orbital-free density functional theory,
$$ E_0 = \mathrm{min}{n} \left[ T{W}[n] + \int \mathrm{d}\mathbf{r} , v(\mathbf{r}) n(\mathbf{r}) \right] \quad\text{with}\quad \int \mathrm{d}\mathbf{r} , n(\mathbf{r}) = 1. $$
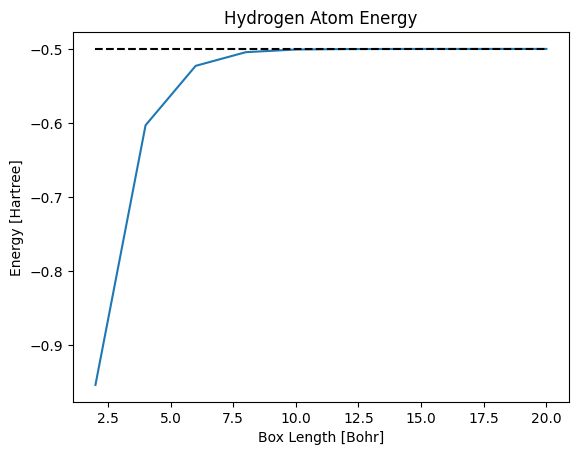
The following code implements this approach, showing that the energy indeed approaches $-\frac{1}{2}$ when the simulation box is sufficiently large.
planewave_cutoff_energy = 50
box_lengths = np.linspace(2,20,10)
energies = []
for box_length in box_lengths:
system = (
profess.System.create(box_length*np.identity(3), planewave_cutoff_energy)
.add_coulomb_ions(1.0, [[0,0,0]], cutoff=0.5*box_length)
.add_electrons()
.add_weizsaecker_functional()
.add_ion_electron_functional()
)
system.minimize_energy()
energies.append(system.energy('h'))
plt.plot(box_lengths, energies)
plt.plot(box_lengths, -0.5*np.ones(box_lengths.size), 'k--')
plt.title('Hydrogen Atom Energy')
plt.xlabel('Box Length [Bohr]')
plt.ylabel('Energy [Hartree]')
plt.show()
One technical point: rather than the full Coulomb potential, the code uses a truncated version set to zero beyond a cutoff radius. This modification facilitates study of the isolated atom with periodic boundary conditions.
$H_2$ molecule#
The electrons in an $H_2$ molecule share a single spatial orbital, so the Weizsäcker functional again provides the exact noninteracting kinetic energy. However, we must now incorporate electron-electron and ion-ion interactions, and the total system energy is
$$ E_0 = \mathrm{min}{n} \left[ T{W}[n] + E_H[n] + E_{xc}[n] + \int \mathrm{d}\mathbf{r} , v(\mathbf{r}) n(\mathbf{r}) + E_{II} \right] \quad\text{with}\quad \int \mathrm{d}\mathbf{r} , n(\mathbf{r}) = 2. $$
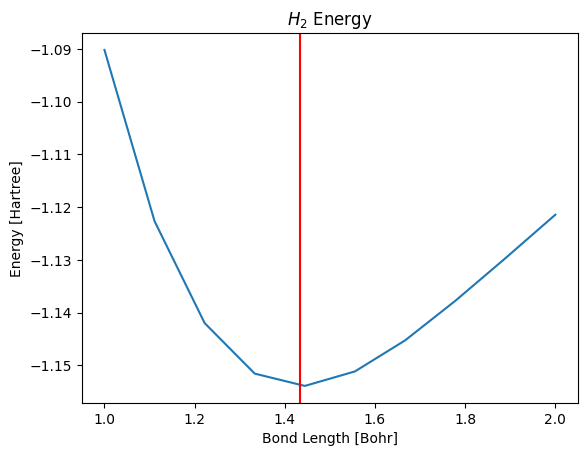
The following code explores the relationship between energy and bond length using the Perdew-Burke-Ernzerhof approximation for the exchange-correlation functional.
system = (
profess.System.create(20*np.identity(3), 20)
.add_coulomb_ions(1.0, [[0,0,0],[1,1,1]])
.add_electrons()
.add_weizsaecker_functional()
.add_hartree_functional()
.add_perdew_burke_ernzerhof_functional()
.add_ion_electron_functional()
.add_ion_ion_interaction()
)
distances = np.linspace(1,2,10)
energies = []
for d in distances:
system.move_ions([[10-d/2,10,10],
[10+d/2,10,10]])
energies.append(system.minimize_energy().energy())
fig, ax = plt.subplots()
ax.plot(distances, energies)
ax.set_title('$H_2$ Energy')
ax.set_xlabel('Bond Length [Bohr]')
ax.set_ylabel('Energy [Hartree]');
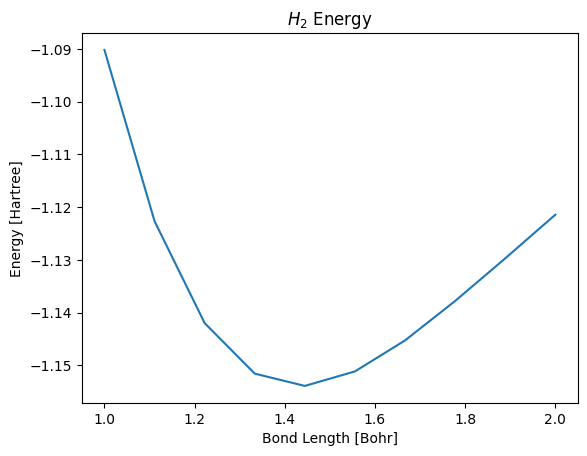
From the plot, it appears the equilibrium bond length is approximately 1.4-1.5 Bohr. To find a precise value, we allow the atoms to relax until all forces are minimized.
ase_tools.minimize_forces(system, 'BFGSLineSearch')
xyz_coords = np.array(system.ions_xyz_coords())
distance = np.sqrt(np.sum((xyz_coords[0,:]-xyz_coords[1,:])**2))
print('\nBond length after minimizing forces: {:4.2f} Bohr\n'.format(distance))
ax.axvline(distance, color='red')
fig
Step[ FC] Time Energy fmax
BFGSLineSearch: 0[ 0] 09:54:14 -1.121424 0.0754
BFGSLineSearch: 1[ 5] 09:54:29 -1.152840 0.0279
BFGSLineSearch: 2[ 7] 09:54:36 -1.153926 0.0030
BFGSLineSearch: 3[ 8] 09:54:38 -1.153936 0.0005
BFGSLineSearch: 4[ 9] 09:54:39 -1.153937 0.0001
Bond length after minimizing forces: 1.43 Bohr