Aluminum#
For face-centered cubic (FCC) aluminum, the build stage might appear as follows.
# choose box vectors and a planewave energy cutoff
box_vectors = 4.05 * np.identity(3)
energy_cutoff = 1200
# create the system
system = profess.System.create(box_vectors, energy_cutoff, ['a','ev'])
# add ions and electrons
system.add_ions(
'potentials/al.gga.recpot',
box_vectors[0,0] * np.array([(0.0,0.0,0.0),
(0.5,0.5,0.0),
(0.5,0.0,0.5),
(0.0,0.5,0.5)]),
'a')
system.add_electrons(system.total_ion_charge())
# add energy terms
(
system
.add_wang_teter_functional()
.add_hartree_functional()
.add_perdew_burke_ernzerhof_functional()
.add_ion_electron_functional()
.add_ion_ion_interaction()
)
The usual next step is to modify the system with minimize_energy(),
finding the ground state electron density and energy.
system.minimize_energy()
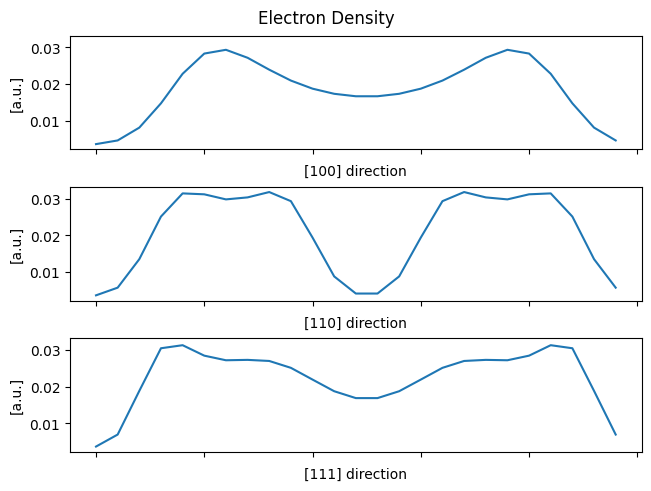
One can then print system properties, inspect the electron density, and more.
print('Volume (A3): {:5.2f}' .format(system.volume('a3')))
print('Grid shape: {:}' .format(system.grid_shape))
print('Total energy (eV): {:5.3f}' .format(system.energy('ev')))
Volume (A3): 66.43
Grid shape: [25, 25, 25]
Total energy (eV): -228.733
fig, ax = plt.subplots(3, 1, constrained_layout=True, sharey=True)
fig.suptitle('Electron Density')
ax[0].plot([system.electron_density[i,0,0] for i in range(system.grid_shape[0])])
ax[1].plot([system.electron_density[i,i,0] for i in range(system.grid_shape[0])])
ax[2].plot([system.electron_density[i,i,i] for i in range(system.grid_shape[0])])
for i, label in enumerate(('[100]', '[110]', '[111]')):
ax[i].set_xlabel(label + ' direction')
ax[i].xaxis.set_ticklabels([])
ax[i].set_ylabel('[a.u.]')
plt.show()